
Faịlụ:Phylogenetic tree of coronaviruses.jpg
Nke káchá mmá na ányá àdíghị.
Phylogenetic_tree_of_coronaviruses.jpg (571 × 451 pixel, ívù akwukwo orunótu: 75 KB, MIME nke: image/jpeg)
{kind=link}
Mmẹkụwátá
| Nkówá |
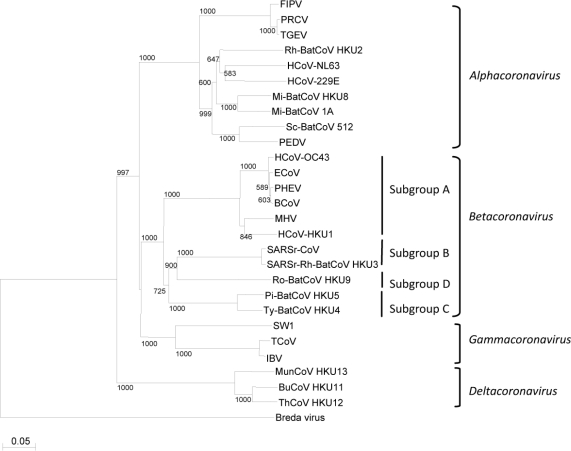
English: Phylogenetic analysis of RNA-dependent RNA polymerases (Pol) of coronaviruses with complete genome sequences available. The tree was constructed by the neighbor-joining method and rooted using Breda virus polyprotein. Bootstrap values were calculated from 1000 trees. 1118 amino acid positions in Pol were included. The scale bar indicates the estimated number of substitutions per 20 amino acids. All abbreviations for the coronaviruses were the same as those in Figure 1. |
| Ǹgụ́ụ̀bọ̀chị̀ | |
| Mkpọlọ́gwụ̀ | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ |
| Odé ákwụ́kwọ́ | Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen |
Nkwényé
Usòrò nke á di okpúrù ákwúkwó íwú nke Mmẹ Òkike Tu ụta Ñkịtị édé íwú nke Í kpó áhà nke ádịghị na úlò 3.0.
- I wepulara nóru:
- i nye – ikọpị,ikekasi na izịpụ ọru a
- i dowaria – igbanwee ọrụ a
- Ọ ga bụ na ọnọdụ ndi a:
- í-kpó-áhà – Ị ga-enyerịrị ugo kwesịrị ekwesị, nye njikọ na ikikere ahụ, ma gosikwa ma emere mgbanwe. Ị nwere ike ime ya n'ụzọ ezi uche ọ bụla, mana ọ bụghị n'ụzọ ọ bụla na-egosi na onye nyere ikikere kwadoro gị maọbụ ojiji gị.
Ịta nke usòrò
Bìri èhì/ogè k'ị hụ òtù ụ̀fa dị̀ m̀gbè ahụ̀.
| Èhì/Ogè | Mbọ-aka | Ógólógó na asaá | Òjìème | Nkwute | |
|---|---|---|---|---|---|
| dị ùgbu â | 02:42, 7 Maachị 2020 | | 571 × 451 (75 KB) | Guest2625 | Uploaded a work by Patrick C. Y. Woo, Yi Huang, Susanna K. P. Lau, and Kwok-Yung Yuen from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3185738/ with UploadWizard |
Ojiji faịlụ
Ihe ndị na-eso ihe eji Ihu akwụkwọ eme na faịlụ a:
Ejiji failụ zụrụ ọha
Wikis ndi a edeputara na eji kwa failụ a:
- Ihe eji na bn.wikipedia.org
- Ihe eji na en.wikipedia.org
- Ihe eji na fa.wikipedia.org
- Ihe eji na fr.wikipedia.org
- Ihe eji na sq.wikipedia.org
- Ihe eji na su.wikipedia.org
{kind=link}